
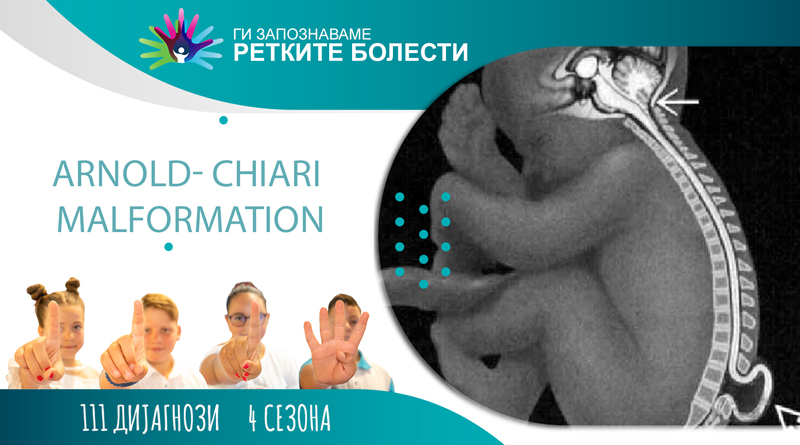
Арнолд-Кјариева малформација
Арнолд-Кјариева малформација / Arnold-Chiari malformation
MKБ-10: Q 07.0
Преваленца/распространетост: непозната
Кјариевата малформација, или попозната како Арнолд–Кјариева малформација (АКМ) (Arnold-Chiari malformaction), претставува развојна структурна аномалија на базата на малиот мозок. Се карактеризира со херниација на тонзилите на малиот мозок во ‘рбетниот канал. Почеста е кај жените за три пати повеќе отколку кај мажите. Преваленцата е 1/1.000 новороденчиња, но се смета дека бројот е поголем, со оглед на тоа дека голем дел од случаите се асимптоматски, или со поблаги симптоми и остануваат недијагностицирани. Зголемениот број на дијагностицирани пациенти се должи на развивањето на МРИ технологијата. Оваа малформација често пати е пропратена со сирингомиелија, при што се формираат и цисти во ткивото на ’рбетниот мозок, пропратени со невролошки појави. Своето име го добила во чест на австрискиот патолог, д–р Ханс Кјари кој првпат ги опишал типовите 1, 2 и 3, а сознанијата за тип 2 ги надополнил неговиот германски колега , д–р Јулиус Арнолд.
Причини:
Точната причина за Арнолд–Кјариева малформација е непозната. Се смета дека причината е структурна аномалија на малиот мозок и ’рбетниот столб. Овој тип аномалии се развиваат за време на феталниот развој. Поради генетски мутации или недостаток на одредени нутритиенти кај мајката за време на бременоста, коскениот простор на базата на черепот е абнормално мал, додека малиот мозок е со нормална големина. Како резултат на тоа, доаѓа до компресија (притисок) на малиот мозок, па циркулацијата на цереброспиналната течност (течноста што циркулира околу мозокот и ‘рбетниот мозок) е блокирана. Поголемиот дел од малформациите се развиваат за време на феталниот развиток, а мал процент можат да се јават подоцна, најчесто како резултат на повреда, инфекција и изложеност на токсични супстанции. Постојат многубројни извештаи во медицинската литература на семејства каде повеќе од еден член во семејството е заболен од Арнолд–Кјариева малформација. Ова сугерира дека во некои случаи генетскиот фактор игра улога во развивањето на оваа малформација. Арнолд–Кјариевата малформација се јавува како дел од поголеми синдроми како Ехлер–Данлос синдром, Голденхар синдром, Олбрајт хередитарна остеодистрофија.
Симптоми:
Симптомите варираат помеѓу пациентите. Има пациенти кои се асимптоматски и се дијагностицираат случајно при некои радиографски испитувања, додека кај други пациенти се јавуват сериозни манифестации на невролошки дефицит.
Најчест симптом поврзан со Арнолд–Кјариева малформација е окципиталната главоболка. Оваа главоболка се јавува во задниот дел од главата со карактер на ширење и предизвикува болки во вратот и рамената. Главоболката ја опишуваат како остра и пулсирачка. Истата може да биде предизвикана или влошена при кивање, кашлање или напрегање.
Други симптоми коишто се јавуваат се: диплопија (двојно гледање), фотофобија (преосетливост на светлина), поматен вид, нистагмус, вртоглавици, тинитус (зуење во ушите), слаба координација или проблеми со рамнотежата, мускулна слабост, дисфагија (отежнато голтање), отежнато зборување, палпитации, синкопа, парестезија, апнеа, и сл.
Сирингомиелијата е често пати поврзана со мноштво од симптомите, зависно од големината и локацијата. Потенцијални симптоми се: губење на мускулната маса, мускулна слабост, губење или намалување на чувствителноста, сколиоза, инконтенција (неконтролирање на сфинктерите), хронична болка, мускулни контракции, некоординирани движења (атаксија) и спазам на мускулите на нозете.
Дијагноза:
Дијагнозата се поставува врз база на симптомите и радиолошко проследување, односно врз база на МРИ технологијата. Обичните рендген снимки и компјутерската томографија не се доволни за поставување на дијагнозата. Најчесто дијагнозата се поставува случајно, откако пациентите се испраќаат на МРИ после подолготрајни проблеми со главоболки, вртоглавици, болки и вкочанетост во екстремитетите и вратот.
Типови на Арнолд-Кјариева малформација:
Најчести типови се 1 и 2. Тип 1 се манифестира кај возрасни (во дваесеттите и триестеттите години) или во подоцнежно детство. Овој тип е најчест и е главен причинител на сирингомиелија.
Тип 2 е посериозна состојба од тип 1 и се манифестира во раното детство. Се карактеризира со хернијација од поголем обем на малиот мозок во ‘рбетниот канал. Може да предизивика живото–загрозувачки копликации за време на раното детство. Често пати е пропратен со спина бифида.
Третман:
Асимптоматските случаи не се третираат, но редовно се контролираат за да се види дали нарушувањето е во прогрес. Симптоматските случаи се решаваат хируршки, најчесто со декомпресија на задната мозочна јама и тоа е единствениот третман за да се коригира функционалниот дефект или да се спречи некоја напредна фаза. Целта на операцијата е да се ослободи притисокот врз мозокот и ‘рбетниот столб и да се овозможи нормален проток на цереброспиналната течност. Операцијата не е лек, туку превентивен зафат со цел да се спречи понатамошно влошување на состојбата.
Некогаш краниектомијата не е доволна, па потребно е и отстранување на дел од првиот вратен пршлен (ламинектомија) и ремоделација на надворешната мозочна обвивка (дуропластика) за ослободување повеќе простор. Хируршкиот третман на Арнолд–Кјариева малформација е со варијабилен успех. Кај некои пациенти после оперцијата има подобрување и олеснување на симптомите, кај други пак се појавува резидуалната болка, мускулната слабост и губење на сензибилтетот.
Во повеќето случаи после декомпресијата, сирингомиелијата се подобрува, или останува со истите димензии. Со тоа се подобрува циркулацијата на цереброспиналната течност, а тоа придонесува за нејзино намалување. На тој начин се олеснуваат симптомите. Кај голем дел од пациентите, за жал, потребно е повеќе од еден оперативен зафат. Во лечењето на Арнолд–Кјариева малформација вклучен е мултидисциплинарен тим составен од невролози, физијатри, педијатри и офталмолози, со цел ублажување и третирање на симптомите.
WebOhrid / 05.02.2022 / Кампања „Ги запознаваме ретките болести“


