
Беквит-Видеман синдром
Беквит-Видеман синдром
Beckwith-Wiedemann syndrome (BWS)
МКБ: Q 87.3
Преваленца: 1-5/10.000
Синдромот Беквит-Видеман (BWS) е генетско нарушување кое се карактеризира со прекумерен раст, предиспозиција за развој на тумор и вродени малформации. Тежината на ова нарушување кај децата варира и обично се препознава при раѓање. Уште во 1963 година, американскиот патолог Беквит го опишал првиот случај на изразена цитомегалија на надбубрежниот кортекс со хиперплазија на бубрезите и панкреасот, што било придружено со хиперплазија на клетките на Лајдиг. Германскиот педијатар Видеман ја дополнил клиничката слика за новата болест со додавање на папочна кила и макроглосијаконски синдром. Синдромот Беквит-Видеман се дијагностицира кај 1 од 10.500 до 13.700 новороденчиња ширум светот, со приближно еднаква инциденца, и кај момчињата и кај девојчињата. Сепак, инциденцата на болеста може да е и повисока бидејќи кај случаите со лесни симптоми, ретко болеста може да биде дијагностицирана.
Генетика на синдромот Беквит-Видеман:
Постојат неколку познати генетски причини за синдромот Беквит-Видеман, кои произлегуваат од промените во експресијата на еден или повеќе гени во регионот 11p15 на хромозомот 11. Генетските причини за овој синдром се комплексни.
Поретко, синдромот на Беквит-Видеман е предизвикан од генетски промени во генот CDKN1C. Овој ген кодира протеин кој го контролира растот на клетките, па доколку овој протеин не работи како што треба, доаѓа до прекумерен раст на клетките.
Наследување на синдромот Беквит-Видеман:
Во околу 85 % од случаите на синдромот Беквит-Видеман во семејството е дијагностициран само еден случај на болеста. Сепак, родителите на дете со овој синдром можат да имаат зголемен ризик за повторување на болеста во која било форма од следните бремености, што зависи од генетската основа на болеста.
Преостанатите 10-15 % од случаите се фамилијарни, и потекнуваат од семејства каде има најмалку еден случај. Во најголем процент од овие семејства, болеста веројатно се наследува автозомно доминантно, со веројатност за повторување од 50 % (1 од 2) за секоја бременост. Во најголем број случаи се работи за генетска промена во генот CDKN1C која е наследена од мајка (генот наследен од таткото е секако инактивиран по пат на метилација или импринтинг).
Поретко, синдромот е резултат на структурна промена во хромозомот 11. Дел од овие промени се случајни, а дел се наследуваат од едниот родител. Ако се наследени, тогаш има определен ризик за повторување на состојбата во следната бременост.
Клиничка слика:
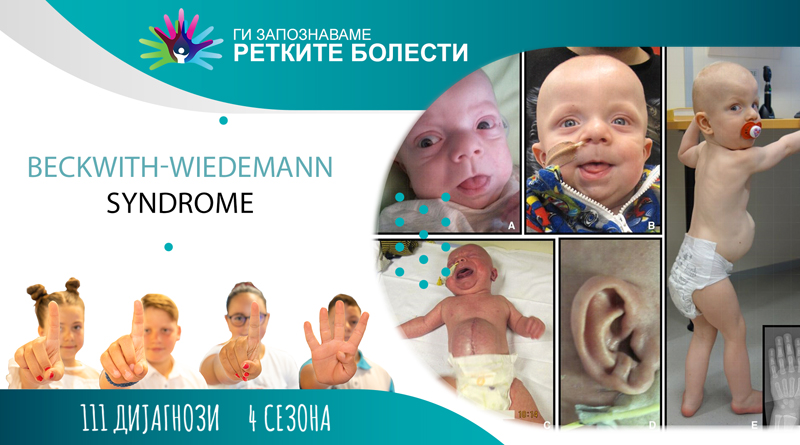
Клиничката слика покажува карактеристична постнатална макрозомија (поголема телесна тежина на новороденчето) и овие деца се повисоки од нивните врсници. Растот забавува околу 8-та година од животот. Кај некои може да се појави абнормален раст само на едната страна од телото. Оваа необична состојба, хемихиперплазија, обично станува помалку изразена со возраста. Повеќето деца имаат дефект на абдоминалниот ѕид (омфалоцела, папочна кила). Исто така, постои макроглосија (зголемен јазик), што може да предизвика проблеми со дишењето, голтањето и говорот, и абнормално големи абдоминални органи – висцеромегалија (црн дроб, бубрези и панкреас).
Децата со овој синдром бараат понатамошен третман, бидејќи имаат зголемен ризик од развој на Вилмсов тумор и хепатобластом. Туморите се развиваат кај околу 10 % од пациентите и речиси секогаш се појавуваат во детството. Хипогликемија (низок шеќер во крвта) се јавува за неколку дена или месеци од животот, и добро реагира на терапијата со дијазоксид. Срцевите малформации се наоѓаат во 9 до 34 % од случаите, а приближно половина имаат кардиомегалија.
Дијагноза:
Дијагнозата се поставува врз основа на клиничката слика. т.е. кога детето ги има сите или некои од карактеристиките на синдромот, и се потврдува со генетско тестирање.
Третман:
Се препорачуваат редовни прегледи за да се открие развојот на потенцијалните карциноми што е можно поскоро.
Ултразвук на абдоменот треба да се прави на секои три месеци до возраст од 7 години. До 4-та година, ултразвукот треба да вклучува прегледи на црниот дроб, бубрезите и другите внатрешни органи. Ризикот од развој на хепатобластом е значително намален кај деца постари од 4 години, така што преостанатите ултразвучни прегледи можат да се фокусираат специјално на бубрезите (ултразвук на бубрезите), кој ги вклучува и надбубрежните жлезди. Ултразвукот на абдоменот е безбеден и безболен, и не вклучува употреба на зрачење.
Нивото на алфа-фетопротеин во крвта треба да се мери на секои три месеци до 4-та година од животот. Овој протеин се ослободува од незрелите или оштетените клетки на црниот дроб, а во високи концентрации се ослободува и од клетките на туморот на хепатобластом. Ова е исклучително чувствителен начин за откривање на овој карцином.
Мултидисциплинарен пристап е потребен во третманот на деца со овој синдром, и може да вклучува: ортопедија, пластична хирургија, ендокринологија и дополнителни потреби кои се засноваат на клиничките проценки.
WebOhrid / 23.02.2022 / Кампања „Ги запознаваме ретките болести“


